
Validação de Sistemas Computadorizados – GAMP5
A validação comprova documentalmente que os sistemas computadorizados cumprem adequadamente com suas funções automáticas e contribuem para garantir a rastreabilidade dos lotes produzidos.
A principal fonte de “inspiração” para os profissionais que validam os sistemas é o guia GAMP5 (Good Automated Manufacturing Practice) – Boas Práticas de Manufatura Automatizada que está atualmente na versão 5 e trás como eixo central, a estratégia de validar baseado em risco (A Risk Based Approach to Compliant GxP Computerized Systems).
GxP é um termo geral para aplicação de boas práticas (good practices). O ‘x’ indica que as boas práticas podem ser relacionadas a qualquer área (fabricação, distribuição, pesquisa clínica, laboratório, etc.). O Guia de Validação de Sistemas Computadorizados da ANVISA que tem o GAMP5 como principal referência e traduz o termo GxP como BPx relevante.
O sistema BPx relevante é todo e qualquer sistema que tem impacto:
- Saúde do paciente;
- Qualidade do produto;
- Integridade de dados.
Normalmente, o impacto na integridade de dados está principalmente relacionado à integridade de dados de rastreabilidade de dados de lote de produção (dependendo do sistema foco da validação). Enfim, todos os sistemas computadorizados que tem relação direta ou indireta com a produção do medicamento/produto para saúde ou impacto na rastreabilidade, tem que ser validado.
Conformidade com FDA 21 CFR Part11
Norma do FDA que estabelece regras para utilização de registro eletrônico nas indústrias de Ciências da Vida. No Brasil, as regras passaram a ser adotas quando a ANVISA publicou a RDC17/2010 onde constam os principais requisitos da norma.
De forma bastante resumida, o sistema deve contemplar:
- Arquivos eletrônicos invioláveis (banco de dados e arquivo protegido);
- Audit Trail – quem entrou no sistema, quando, o que fez, porque fez, de onde fez?
- Controle de acesso ao sistema – níveis de senhas de usuários e privilégios;
- Assinatura eletrônica = ID + senha;
- Políticas de usuários aposentados;
- Garantia de uso de contas por seus usuários genuínos;
- Controle rigoroso de recuperação de senhas;
Os requisitos do FDA 21 CFR Part11 são atualmente bastante comuns no mercado e se previstos desde o início do projeto, traz a segurança e rastreabilidade necessária para o bom uso do sistema.
Para sistemas legados que não atendem o regulamento, o melhor caminho é a Análise de Riscos. Sugerimos estudar justamente a ausência destas seguranças eletrônicas. Se o risco resultar nos níveis ‘médio’ e ‘alto’, uma medida de mitigação deve ser prevista. Caso a mitigação ou upgrade não seja possível, a troca do sistema deve ser considerada.
Como validar?
O GAMP5 foi publicado em 2008 e desde então revolucionou o método de validação de sistemas. A melhor forma de validar é sem dúvida, baseada em riscos, seja um sistema novo (validação prospectiva) ou sistema legado (validação concorrente).
A primeira atividade importante do profissional de validação é reunir toda a documentação disponível sobre o sistema objeto do estudo. Em seguida, este profissional passa a estudar a documentação com o objetivo de identificar cenários de riscos.
Em equipe multidisciplinar, para cada cenário de riscos levantado, são analisados itens utilizando o seguinte fluxo de trabalho:
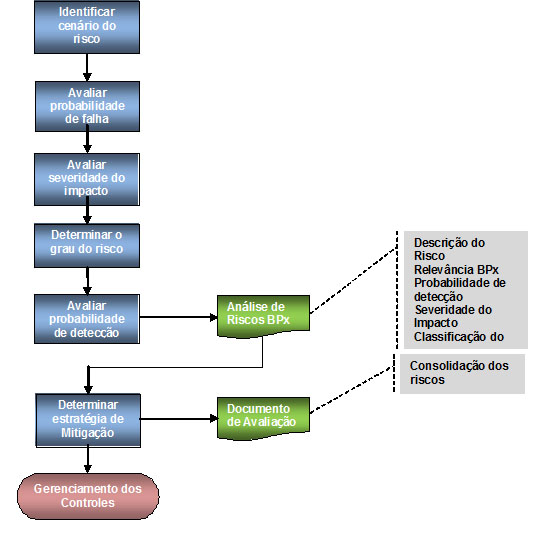
O estudo da documentação existente para levantamento dos cenários de riscos é de extrema importância para conhecer as particularidades do sistema a ser validado. Entretanto, após alguns sistemas validados pelo profissional, alguns cenários de riscos são invariavelmente apresentados, como comportamento do sistema perante queda de energia, estudo dos perfis de acesso, segurança de acesso, qualidade do registro eletrônico produzido pelo sistema incluindo trilha de auditoria, aplicação de assinaturas eletrônicas, etc.
É importante ressaltar que a Análise de Riscos tem que ser feita por equipe multidisciplinar porque é muito comum a necessidade de adoção de medidas de mitigação para os riscos que resultam em níveis ‘médio’ e ‘alto’. A adoção destas medidas tem que ser acordada entre a equipe para que realmente funcionem, sejam elas adoção de novos procedimentos de trabalho ou melhorias no sistema.
As medidas a serem adotadas se tornam a estratégia de validação e são os principais pontos onde a validação deve focar. Por isso, o GAMP5 preconiza a validação baseada em riscos. Normalmente, estas medidas são detalhadas na URS (User Requirement Specification) que passa a ser o documento referência da validação.
Se o sistema for de prateleira, basicamente devem ser elaborados testes que comprovam que os requisitos da URS foram cobertos e alguns outros testes típicos de sistemas cobertos nas fases de:
- Qualificação de Instalação;
- Qualificação de Operação;
- Qualificação de Desempenho.
Se o sistema for configurado ou customizado para atender as necessidades da empresa usuária, documentos de especificações devem ser produzidos. Alguns exemplos:
- Especificação Funcional;
- Hardware Design;
- Software Design.
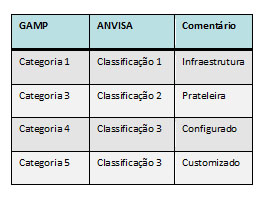
Tanto o GAMP5, como o Guia da ANVISA detalham o ciclo de vida exato necessário para cada tipo de sistema e separam da seguinte forma:
Para esclarecer melhor, o fluxo a seguir mostra os documentos que devem ser previstos para a validação de um sistema ou equipamento de fábrica, configurado, classificação 3 da ANVISA. Neste exemplo, os documentos de especificação e protocolos de testes fariam parte do escopo do fornecedor:
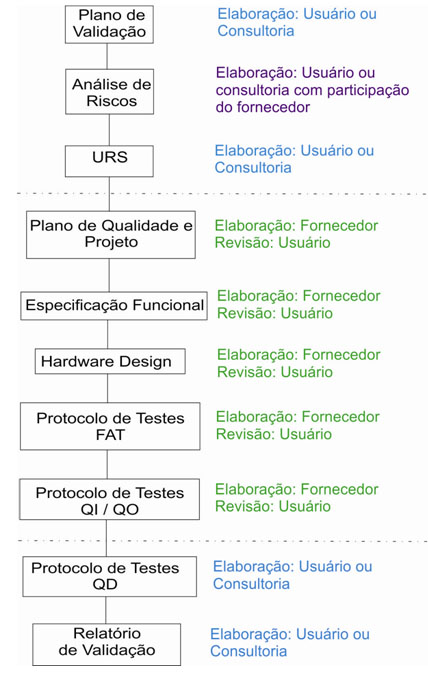
É importante ressaltar que adquirir o pacote de documentos do fornecedor não reflete a conclusão do trabalho de validação. É necessário “Abrir” o ciclo de vida de documentos, com emissão do Plano de Validação, Análise de Riscos e URS e “Fechar” o ciclo de vida com a emissão do protocolo de testes de Desempenho, Matriz de Rastreabilidade e Relatório de Validação, documentos que normalmente não faz parte do escopo do fornecedor da solução.
Conformidade com FDA 21 CFR Part11
Norma do FDA que estabelece regras para utilização de registro eletrônico nas indústrias de Ciências da Vida. No Brasil, as regras passaram a ser adotas quando a ANVISA publicou a RDC17/2010 onde constam os principais requisitos da norma.
De forma bastante resumida, o sistema deve contemplar:
- Arquivos eletrônicos invioláveis (banco de dados e arquivo protegido);
- Audit Trail – quem entrou no sistema, quando, o que fez, porque fez, de onde fez?
- Controle de acesso ao sistema – níveis de senhas de usuários e privilégios;
- Assinatura eletrônica = ID + senha;
- Políticas de usuários aposentados;
- Garantia de uso de contas por seus usuários genuínos;
- Controle rigoroso de recuperação de senhas;
Os requisitos do FDA 21 CFR Part11 são atualmente bastante comuns no mercado e se previstos desde o início do projeto, traz a segurança e rastreabilidade necessária para o bom uso do sistema.
Para sistemas legados que não atendem o regulamento, o melhor caminho é a Análise de Riscos. Sugerimos estudar justamente a ausência destas seguranças eletrônicas. Se o risco resultar nos níveis ‘médio’ e ‘alto’, uma medida de mitigação deve ser prevista. Caso a mitigação ou upgrade não seja possível, a troca do sistema deve ser considerada.
Vantagens da Validação
Além de contribuir para a qualidade e integridade de dados, o processo de validação é valioso, pois, permite:
- Extrair todos os recursos disponíveis no sistema necessário para cobrir de forma segura o processo em específico;
- Tornar a equipe detentora de conhecimento técnico mais aprofundado sobre o sistema, evitando que o conhecimento fique inteiramente nas mãos do fornecedor (abertura da “caixa preta”);
- Documentar as descobertas técnicas, evitando-se a perda do conhecimento na eventual saída dos profissionais da empresa (risco ao negócio!!);
- Explorar todos os recursos automáticos possíveis de forma a evitar a etapa manual de produção, colaborando para que o produto tenha repetibilidade e reprodutibilidade (objetivos da validação de processos!!);
- Direcionar a equipe analisar ações necessárias e procedimentos documentados para plano de contingência, backup de dados e de aplicação e recuperação de desastres, diminuindo tempo de parada de produção (risco ao negócio!!);
- Confiabilidade nas informações de processo evitando-se erros operacionais.
Em alguns setores da indústria nacional, como por exemplo, o farmacêutico e o farmoquímico a validação de sistemas computadorizados é uma exigência regulatória, e uma tendência em outros, como a indústria cosmética e a veterinária.
Mesmo que a legislação nacional não exija a validação de sistemas computadorizados para algumas indústrias, quando a intenção é exportação, existem vários países onde a validação é obrigatória para determinados segmentos, como é o caso da indústria veterinária.
Glossário
FAT: Factory Acceptance Test / Teste de Aceitação de Fábrica;
FDA: Food Drugs and Administration (Agência Reguladora dos Estados Unidos);
CFR: Code of Federal Regulation – regras gerais e permanentes vigentes no governo federal dos Estados Unidos;
ID: Abreviação da palavra ‘identificação’;
QI: Qualificação de Instalação;
QO: Qualificação de Operação;
QD: Qualificação de Desempenho.
Sílvia Martins e João Gomes – consultores da empresa Five Validação de Sistemas Computadorizados.



Esta matéria foi muito enriquecedora.
E qual categoria as planilhas eletrônicas devem ser classificadas e qual é o tratamento em relação da validação dessas planilhas.
Grato
Parabéns ! Excelente artigo.